
在新冠疫情期间,美国政府花费了数十亿美元购买了近 400 种产品,旨在保护、诊断和治疗数亿人,所有产品都带有“EUA”或“紧急使用授权”标签。
但 EUA 的实际含义是什么?
甚至在我们回答这个问题之前,通过了解 EUA 相对于授权或批准医疗产品的其他途径的地位,了解一下是有帮助的: EUA 不是什么:
EUA 不是正在进行临床试验的实验产品的指定
如果我们只了解 EUA 的一件事,那就应该是:EUA 不适用于正在进行受 FDA 法规或其他法律要求管辖的临床试验的产品。
EUA 也不同于扩展使用(EAU),通常称为“同情使用”使用,它适用于在实验产品获得完全批准之前向患有严重、不治之症的患者提供使用实验产品的机会。
这张表来自一个 FDA-CDC 2020 演示 总结了正在进行临床试验的产品、通过扩大“同情”访问范围向患者提供的产品以及通过 EUA 授权的产品之间的差异:
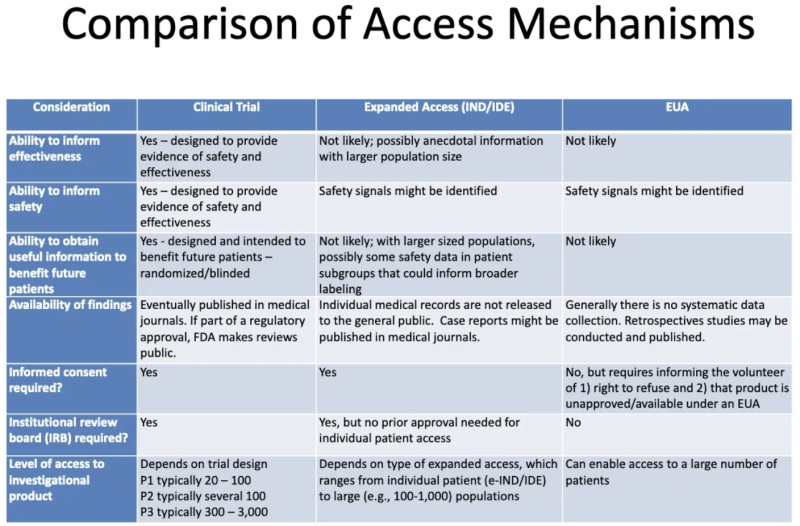
该表告诉我们有关 EUA 的信息:
- 授予 EUA 的过程不太可能生成有关产品有效性的任何信息。
- 授予 EUA 的过程并不是为了提供安全性或有效性的证据,但可能会识别安全信号。
- 一旦产品获得 EUA 并向某些患者施用,就不太可能获得任何有用的信息来使任何未来的患者受益。
- 没有关于 EUA 有效性或安全性的系统数据收集,并且作为监管审批流程的一部分,也没有在医学期刊上发布任何数据。
- 无需知情同意,但必须告知“自愿”服用该产品的患者他们可以拒绝,并且该产品未经 EUA 批准/可用。
- 不需要机构审查委员会 (IRB)。 [IRB 是一个旨在保护临床试验中人类受试者福祉的委员会]
为了进一步澄清 EUA 与任何正常审批流程的区别, 2009 年美国国家科学院医学研究所出版物,我们发现这样的说法:
重要的是要认识到 EUA 不是开发路径的一部分;它是一个完全独立的实体,仅在紧急情况下使用,并且不属于药品审批流程的一部分。 (第 28 页)
总结如下:
授予产品 EUA 的过程不太可能产生任何安全性或有效性的证据。一旦产品获得 EUA 并向患者施用,就不太可能获得任何有用的信息来使未来的患者受益,因为没有关于有效性或安全性的系统数据收集。
根据 CDC/FDA 和 IMNA 提供的所有这些非常明确的信息,可以公平地得出结论,紧急使用授权是一个应该非常明智地应用的过程,并且仅在紧急情况下应用。
现在让我们看看 EUA 在法律上旨在解决哪些类型的紧急情况。
EAU 旨在应对大规模杀伤性武器紧急情况
上述允许 EUA“准入机制”的法律是针对涉及大规模杀伤性武器(WMD)(也称为 CBRN(化学、生物、放射性、核)剂)的极端、紧急紧急情况而制定的。
食品药品监督管理局是这样处理的 (FDA) 描述其 EUA 权力:
FD&C 法案第 564 条(21 美国法典360bbb–3)允许 FDA 加强针对生物、化学、核和放射性制剂的公共卫生保护。
凭借这一 EUA 授权,FDA 可以帮助确保在没有足够的、经批准的医疗对策的情况下,在紧急情况下可以使用医疗对策来诊断、治疗或预防由生物、化学、核或放射性制剂引起的严重或危及生命的疾病或病症。 ,以及可用的替代方案(以及其他标准)。
这些 EUA 权力于 2004 年在与 CBRN 特工攻击准备相关的非常特殊的情况下被授予。
如前所述 哈佛法健康法案,
最终,正是反恐战争导致了紧急使用授权。 11 年 2001 月 XNUMX 日事件和随后的炭疽邮件攻击之后,国会颁布了 2004 年生物盾计划法案.
记录 表明国会特别关注生物恐怖的威胁,而不是为自然发生的流行病做准备。
鉴于涉及大规模杀伤性武器攻击的此类真正极端紧急情况,EUA“准入机制”不需要大量监管或遵守任何制造或临床试验标准,这是可以理解的。
那么EUA准入机制到底需要什么?
紧急使用授权 (EUA) 的 3 个步骤
为了向医疗产品授予 EUA,必须满足三件事:
- 国土安全部部长、国防部长或卫生与公共服务部部长需要确定是否存在涉及 CBRN 制剂攻击或攻击威胁或由此类制剂引起的疾病的紧急情况。
- FDA 在发布 EUA 时需要确保其符合四项“法定标准”。
- FDA 必须在 EUA 中“施加某些必需的条件”。
EUA 第 1 步:宣布 CBRN 紧急状态
EUA 的紧急声明是独立的,与总统、HHS 部长或其他任何人可能发布的任何其他紧急声明无关。它必须专门为了激活 EUA 而发布,并且可以独立于任何其他紧急声明而结束或延长。
下面是 EUA 法律规定 激活 EUA“准入机制”有四种可能的情况:
- 国土安全部部长确定存在国内紧急情况或极有可能发生国内紧急情况,涉及生物、化学、放射性或核制剂攻击的较高风险;
- 国防部长确定存在军事紧急情况,或很可能发生军事紧急情况,涉及较高的风险 联合的 州 军事力量,包括在第 10 章或第 50 章授权下行动的人员,使用以下方式进行攻击:
- 生物、化学、放射或核制剂;或者
- 可能对美联航造成迫在眉睫的生命威胁和特定风险或与之相关的代理人 州 军事力量;
- 的决定 董事会秘书 [卫生与公众服务部]存在公共卫生紧急情况,或极有可能发生公共卫生紧急情况,影响或极有可能影响国家安全或人民的健康和安全 联合的 州 居住在国外的公民,涉及生物、化学、放射或核制剂,或可能由此类制剂引起的疾病或病症;或者
- 根据《条例》第 319F-2 条识别重大威胁 公共卫生服务法 [42 美国法典247d–6b] 足以影响国家安全或人民的健康和安全 联合的 州 居住在国外的公民。
EUA 第 2 步:满足法定标准
一旦其中一位秘书宣布存在需要紧急使用授权的紧急情况,FDA 还必须满足四个“法定标准”才能颁发紧急使用授权。 FDA 对这些要求的解释如下:
- 严重或危及生命的疾病或状况
为了让 FDA 发布 EUA,HHS 部长 EUA 声明中提到的 CBRN 制剂必须能够引起严重或危及生命的疾病或状况。
- 有效性的证据
可以考虑获得 EUA 的医疗产品是那些“可能有效”预防、诊断或治疗严重或危及生命的疾病或病症的产品,这些疾病或病症可能由 HHS 部长声明中确定的 CBRN 制剂引起。第 564(b) 条规定的紧急情况或紧急情况威胁。
EUA 的“可能有效”标准提供的证据水平低于 FDA 用于产品批准的“有效性”标准。 FDA 打算使用风险效益分析来逐案评估可能的 EUA 产品的潜在有效性,如下所述。
[添加粗体]
- 风险收益分析
如果专员确定该产品在用于诊断、预防或治疗已确定的疾病或状况时的已知和潜在益处超过该产品的已知和潜在风险,则可以考虑对该产品进行 EUA。
在确定产品的已知和潜在益处是否超过已知和潜在风险时,FDA 打算看看 综合科学证据来做出总体风险收益确定。此类证据, 可能出现 从各种来源, 可能包括 (但不限于):国内外临床试验结果、动物模型体内疗效数据、体外数据, 可供 FDA 考虑。 FDA还将评估产品的质量和数量 现有证据,考虑到科学知识的当前状态。
[添加粗体]
- 别无选择
FDA 要想发布 EUA,必须没有足够的、经批准的和可用的候选产品替代品来诊断、预防或治疗疾病或病症。如果批准的替代品的供应不足以完全满足紧急需求,则潜在的替代产品可能被视为“不可用”。
EUA 步骤 3. 施加所需条件
一旦我们有了针对 EUA 的紧急声明,一旦 FDA 确定该产品可能有效,并且无论有什么证据表明其益处大于风险,就会有更多一层相关监管。
这是如何 2018 年国会研究服务部关于 EUA 的报告 解释一下:
FFDCA §564 指示 FDA 在 EUA 中施加某些必需的条件,并在适当的情况下允许附加酌情条件。所需条件有所不同,具体取决于 EUA 是针对未经批准的产品还是针对已批准产品的未经批准的使用。对于未经批准的产品,使用条件必须:
(1) 确保管理该产品的医疗保健专业人员收到所需的信息;
(2) 确保接受该产品的个人收到所需的信息;
(3) 规定与产品相关的不良事件的监测和报告;和
(4) 规定制造商的记录保存和报告。
结论
正如本文所述,FDA/CDC 明确认识到授予紧急使用授权 (EUA) 的过程不太可能生成有关产品有效性或安全性的任何信息。当我们查看 EUA 的法律条文时,我们发现这确实是一个正确的评估。
EUA 法没有强加任何可能确定产品是否安全或有效的法律或监管标准。唯一的标准是 FDA 是否认为该产品可能有效,以及其已知的好处是否超过其已知的危害。如果没有已知的危害或已知的益处,因为该产品从未经过药物审批流程,FDA 可以使用其选择的任何信息或标准来做出决定。
由此可见,其产品是 EUA 候选产品的公司可能会尝试通过其选择的任何方式证明产品的安全性和/或有效性。这种尝试的存在(无论是临床试验还是其他数据收集机制)以及如何进行这种尝试都取决于公司。 EUA 法律中的任何内容均不适用于公司如何设计、进行或分析其选择追求的任何研究或其他数据收集机制。
应用于 Covid 产品,这意味着:
- Covid 产品无需获得 EUA 即可获得临床试验的安全性或有效性数据。
- EUA 流程中提到的任何临床试验都是在没有法律适用的监管标准的情况下进行的。
- 当我们发现这些产品缺乏功效或安全性时,这并不奇怪。这是该过程极有可能的结果。
- EUA 流程中没有数据可作为有关产品安全性或功效的非 EUA 决策的依据。因此,任何非 EUA 使用该产品都需要从一开始就经过常规医疗产品的法律审批流程。
有关新冠疫苗审批流程的更多信息 此处.
转载自作者 亚组
发表于 知识共享署名4.0国际许可
如需转载,请将规范链接设置回原始链接 褐石研究所 文章和作者。








